J’ai effectué cette semaine en labo la synthèse du ferrocène et de certains de ses dérivés dont l’acétylferrocène. Lors de la réaction, des cristaux rouges sont censés se former lors de la cristallisation. Tout d’abord, j’ai eu très peu de cristaux. Il y a eu des difficultés à cristallisé le produit et lors d’un test sur CCM, j’ai majoritairement encore du ferrocène au lieu de l’acétylferrocène alors que Ac2O était en excès. Pour être exact, j’avais deux signaux: un correspondant au ferrocène et l’autre à l’acétylferrocène. Je précise que j’ai effectué une filtration et évaporé pas mal de solvant sous
pression réduite.
J’ai donc bien le produit désiré mais j’ai pas réussi à l’ "extraire" au vu de la CCM.
Deuxième petite question. Selon vous, à quoi est du la plus grande réactivité du ferrocène par rapport au benzène ? Je pense que ça doit être dû à ce qu’on a le même nombre d’électrons délocalisés sur uniquement 5 atomes (au lieu de 6) et sûrement aussi à cause du $\pi$-backdonation.
Cool ça ! Selon mon protocole, je devais ajouter le ferrocène brut à l’Ac2O mais effectivement j’ai l’impression d’avoir fais l’inverse; j’ai donc ajouté lentement Ac2O au ferrocène avec un peu d’acide phosphorique pour accélérer tout ça.
Ton protocole était donc comme le mien, ce qui n’a pas de sens :
Lorsque tu ajoute du ferrocène dans une plein pot de $Ac_2O$ tu met en contact 1 molécule de $FeCp_2$ et plein de $Ac_2O$
A ce moment là il y a, dans un court instant, assez de $Ac^+$ pour faire des $SEAr$ à plusieurs reprises (même si je te l’accorde ça se désactive à partir de 3 acylation).
Tu as plus de chance d’obtenir 2 Cp acylé qui entoure ton ion fer.
Alors que si tu fais l’inverse… Tu es dans l’ordre logique des choses, soit sure d’avoir suivi l’une ou l’autre méthode
EDIT : pour la grande réactivité par rapport au benzène, c’est qu’une charge négative c’est moins stable qu’un objet neutre; donc en soit un cyclopentadienure c’est plus nucléophile qu’un cyclopentadiène donc surement plus qu’un benzène. Tu as le même nombre d’electron disponible sur une plus petite surface, densité électronique accrue
Ton explication fait sens. Par contre, expérimentalement j’ai des amis qui ont suivi le protocole et la double acylation ne se voyait pas sur le spectre RMN.
Selon moi, le problème que j’ai eu c’est que mon ferrocène était encore assez mouillé quand je l’ai mis. La raison pour laquelle ça n’a pas cristallisé je suppose que c’est parce que la concentration d’acetylferrocène n’était pas assez élevée?
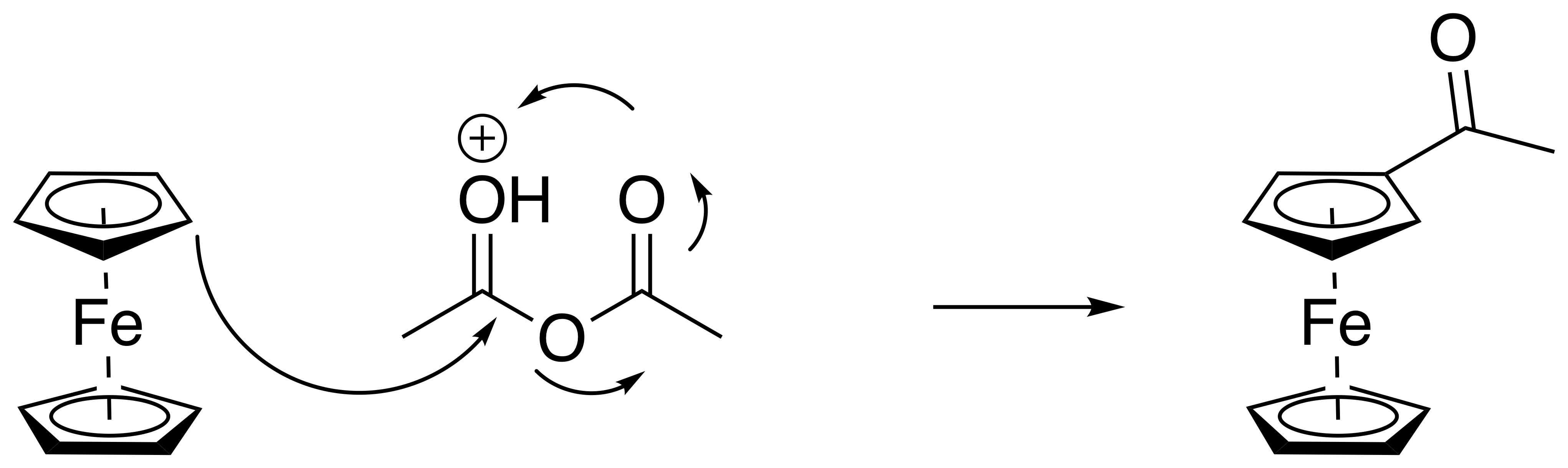
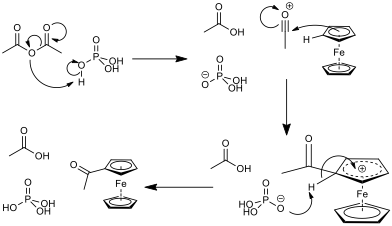
D’ailleurs, concernant le mécanisme, tu penses que c’est bien celui-ci ? Je l’aurais personnellement pas fait passé par cet intermédiaire.
Ah tiens, tu as tout as fait raison. cela se joue sur un truc que dont je ne me rappelais plus. En fait c’est l’oxygène $\text{sp}^2$ qui est plus basique que l’oxygène pontant l’anhydride. Donc ton dessin est meilleur excuse moi.
Tiens j’ai mon cahier sous la main j’vais voir ça en detail j’edit
EDIT : Je décide de ne t’aider que sur le rendement, j’suis pas un fifou de l’analyse. Pour être tout a fait honnête ma réaction c’était avec : $\text{AlCl}_3$ et $\text{Me-COCl}$ donc on est pas dans les même conditions opératoires.
Combien avais-tu d’eq. de $H_3PO_4$ ?
Comme tu joue sur l’acidité d’un oxygène $sp^2$, et que tu fabrique un $Cp-CO-Me$ ce nouvel oxygène peut aussi capter ton $H^+$. Donc c’est important
Pour la purification tu as fait comment ?
Je sais que j’ai du y aller à la colonne de chromatographie (c’était jolie à voir) avec un gradient d’éluant de 18% AcOEt dans de l’ether de petrole à 30% AcOEt dans l’ether de petrole.
Avec un $Rf = 0.68$ dans $15/85$ AcOEt/PE.
Notes diverses
89% de rendement mon gars <3
On a fait la réaction sur 1 heure, c’était stipulé plus. Cela peu jouer sur le rendement. On a ajouter le chlorure d’éthanoyle sur le Ferrocène avec l’$AlCl_3$ (2 eq) en solution. Le tout dans le DCM.
J’avais environ 5 équivalents d’acide phosphoriques. J’y ai peu être été un peu fort là-dessus mais j’ai neutralisé la solution par la suite avec de NaHCO3.
Pour la purification, j’ai simplement fais une recristallisation.
Voici ci-dessous plus de détails sur le protocole suivi:
Dans un ballon, le ferrocène est ajouté. Puis, ajout de l’anhydre acétique et ajout de l’acide phosphorique (goutte à goutte). On a chauffé à 90°C pendant environ 45 minutes. Le mélange réactionnel est versé dans de la glace pilée sous agitation afin d’arrêter spontanément la réaction. Quand toute la glace a fondu, on neutralise lentement la solution avec du bicarbonate de sodium jusqu’à être à pH 7. Ensuite, on filtre sur Büchner. On dissout le solide jaune-brun dans de l’ether de pétrole et on ajoute du sulfate de magnésium pour sécher. On filtre de nouveau et on évapore à pression réduite une partie du solvant pour ensuite effectuer une cristallisation. Et finalement, test CCM & point de fusion.
Mhm j’ai plusieurs hypothèses à émettre alors, mais j’met la reserve qu’on ne peu que faire des spéculations, faudrait faire des recherches croisé sur SciFinder pour trouver les vraies réponses s’il y en a :
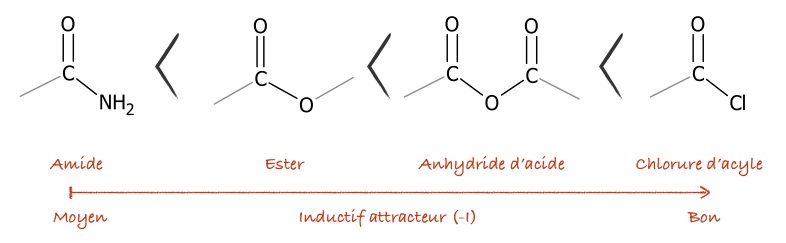
La synthèse via $H_3PO_4$ me semble sympathiquement moins logique de celle au chlorure d’éthanoyle. En fait avec $AcCl$ on as pas un acide en solution qui pourrait faire une réaction critique ruinant l’aromaticité.
$$
H^+ + Cp^- \rightarrow CpH
$$
(On produit du $HCl$ (avec la méthode $AcCl$) mais c’est un gaz donc on peut imaginer qu’il s’évapore de la solution tranquille.)
45 minutes c’est court, la cinétique d’un electrophile comme l’ion acylium produit par un anhydride d’acide n’est paussi bonne que pour un acylium créer par $AcCl$
Pour rappel de réactivité
Alors que mon mode opératoire donnait 1h30 et que j’ai fais 1h de mise en reaction. Ça expliquerait un taux de conversion bas pour ton mode opératoire.
De mon coté on a jamais mis notre milieu reactionnel dans l’eau ou dans un milieu protique ($H_2O$ ou $HCO_3^-$ comme tu l’as fait) donc on as encore une voie parasites à la consommation de $Cp^-$
Par contre, expérimentalement j’ai des amis qui ont suivi le protocole et la double acylation ne se voyait pas sur le spectre RMN.
Étrange, en chromatographie on a observé un produit qui avait un Rf cohérent avec la double acétylation… Le Fer ne pose pas de problème pour la RMN ?! cool.
Selon moi, le problème que j’ai eu c’est que mon ferrocène était encore assez mouillé quand je l’ai mis. La raison pour laquelle ça n’a pas cristallisé je suppose que c’est parce que la concentration d’acetylferrocène n’était pas assez élevée?
Ah bah la recristallisation c’est toujours un peu délicat, faut avoir un peu de doigté… Selon le solvant est la quantité de matière que tu as à recristalliser ça peut être très chaud. Je sais qu’on aime faire appel à la recristallisation quand on a bcp de produit généralement, pour éviter de perdre quelque peu dans les solvants.
Je sais que pas mal d’atome exotique ne passe pas en RMN par rapport à leurs spins, mais j’ai pas regardé sur celui du fer brouillerais ou pas l’analyse
Si jamais tu le veux j’peux te passer un récap de mon mode opératoire. Mais ça risque de t’être inutile
Connectez-vous pour pouvoir poster un message.
Connexion
Pas encore membre ?
Créez un compte en une minute pour profiter pleinement de toutes les fonctionnalités de Zeste de Savoir. Ici, tout est gratuit et sans publicité.
Créer un compte





{kind=link}